At the second exposomics conference hosted by the Icahn School of Medicine at Mount Sinai, the field felt less like a finished discipline than a discipline discovering the shape it may soon take. The word “exposome” has been around for a little more than a decade, but only recently has the area begun to accelerate in a visible way. The easiest indicator is crude but useful: the number of papers. By that measure, exposomics today resembles genomics roughly twenty years ago. The methods are not yet mature, the standards are not yet settled, but the outlines are becoming clear.
In the United States, the National Institutes of Health was among the earliest institutions to promote the concept. UC Berkeley and Emory were also early centers of activity. Mount Sinai founded what was described as the first exposomics institute in the United States in 2017, using the network of American medical schools to push the field forward. The people gathering around the subject now come heavily from medical-school ecosystems, and it is not hard to imagine a surge in both funding and publications over the next decade. At the moment, the field still feels like it is in its angel-investment round.
What exposomics is trying to measure
The central question is close to the one genomics has been asking: what determines health? Genomics tends to put much of the explanatory burden on genes, and with sequencing becoming increasingly affordable at the individual level, that approach has become technically and economically plausible. Exposomics starts from a different discomfort: genes alone cannot explain enough.
A person’s health status may involve genetics, but also epigenetics, the proteome, the metabolome, and everyday exposures. It may also depend on location, socioeconomic status, the gut microbiome, and other contextual variables that do not fit neatly into a single biological layer. Health is the outcome; the predictors are numerous. This is very obviously not a one-factor model.
That is why exposomics is an intensely problem-oriented and highly synthetic field. Its foundations include, but are not limited to, statistics, life sciences, data science, social science, environmental science, analytical chemistry, toxicology, public health, medicine, remote sensing, sensor technology, automation, and information science. It is still unclear which of these will prove most important. What is already obvious is that any one of them can become the weak link in answering the larger question. In practice, almost every discipline involved has its own weak points, and the barriers between disciplines are much higher than people like to admit.
Two of the most immediate problems can be seen from the angles of environmental analytical chemistry and data science.
The analytical chemistry problem: you cannot target what you do not know
If exposure is to be evaluated, the first task is to know what is present. That is the logic of targeted analysis. Unfortunately, exposomics usually begins precisely where targeted analysis becomes insufficient: we do not know in advance what is in the sample.
For that reason, many studies borrow from metabolomics and use high-resolution mass spectrometry to collect information on unknown compounds. The immediate output is a set of chromatographic and mass-spectrometric peaks. But full-scan high-resolution mass spectrometry is messy. It often contains large numbers of adducts, fragments generated by in-source reactions, and isotope peaks of the same compounds. One may collect tens of thousands of peaks at once, while the number of actual compounds producing those peaks may be only a tenth as large. These peaks are also correlated with one another. If peak-level data are used directly to discuss correlations among substances, bias is almost inevitable.
Peak-picking algorithms are another weak point. Many of them behave poorly on full-scan data. Signals that should not be treated as peaks are selected as peaks, and the integration can be terrible. From the standpoint of analytical chemistry, that is not a small technical inconvenience; it is unacceptable if the downstream interpretation depends on those features.
Naming the unknown is harder than detecting it
The next problem is annotation. A common workflow is to run a full scan, screen for differential peaks, and then acquire MS/MS spectra for those peaks. In some cases, the differential peaks are annotated directly.
Gas chromatography–mass spectrometry data can be set aside for the moment, because under hard ionization the fragments are often specific enough to support qualitative identification. Liquid chromatography is a different story. Unless one is using a less mainstream ionization source such as APPI, LC-MS typically relies on ESI or APCI, both of which are soft ionization techniques. In MS1, there is often very little information that is truly useful for confident identification. Using MS1 alone for qualitative assignment is risky, and any downstream pathway analysis built on that assignment becomes unreliable.
Even if an MS1 match is found, isomerism remains unresolved. Isomers can have very different biological activities. On top of that, the major databases are fragmented, each with its own scope and limitations. A unique annotation does not necessarily mean a reliable identification.
MS/MS-based identification can now be done with many software tools, but these tools are still largely underfit for the problem. Their training data depend heavily on available standards or on spectra shared by community users. That makes genuinely unknown compounds extremely difficult to identify.
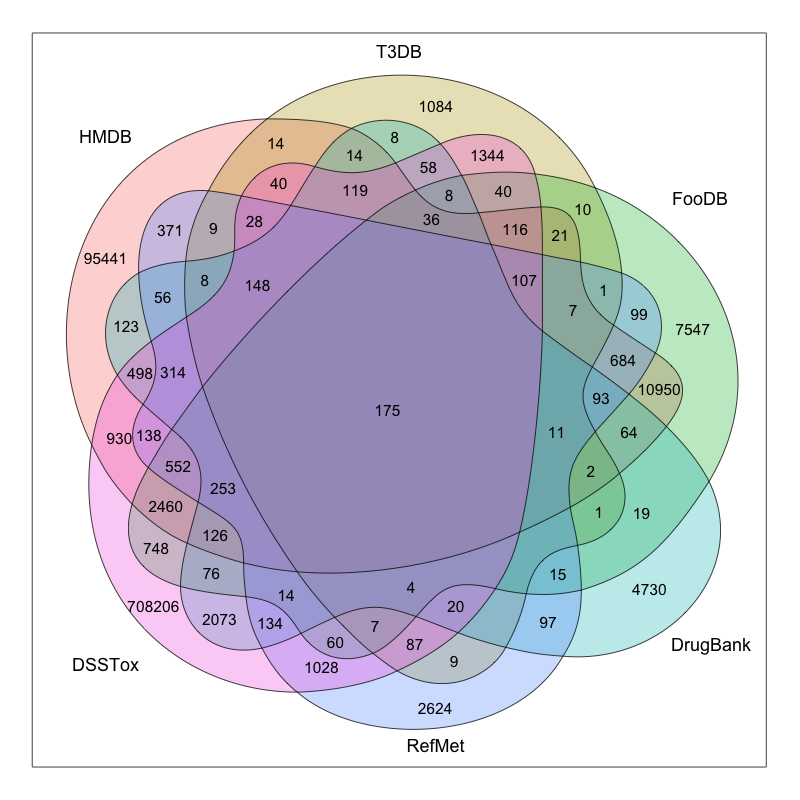
The coverage of current compound databases illustrates the difficulty. The three largest chemical resources—PubChem, ChemSpider, and CAS—are often not easy to integrate into a practical workflow, either because the data are inaccessible or because processing them is extremely laborious. CAS is likely the largest, with about 140 million substances, though the fraction that most researchers can realistically encounter or use is much smaller.
In metabolomics, HMDB is among the most commonly used databases, but from an exposomics perspective much of that content is endogenous. For exogenous bioactive substances, databases such as DrugBank, ChEBI, and T3DB become relevant. For industrial chemicals, resources such as the HPV database may be useful. Yet even these are the relatively fortunate cases where information can be found. Some substances may have little more than an InChIKey associated with them and almost no other usable data.
There are not many places that can systematically collect, harmonize, and curate all of this information. In some databases, the underlying data organization is also poor: formats are inconsistent, fields are irregular, and basic extraction can become confusing unless the user already has substantial domain expertise.
Throughput creates its own batch effects
Analytical throughput is easy to underestimate. Suppose a study has 100 samples and each run takes 30 minutes. After adding quality-control samples, the sequence may grow to about 150 injections. That is three to four days of continuous analysis. During that time, the column ages, and even the mass axis can drift. Calibration can be repeated, of course, but the result is that a clear batch effect may appear even within what is nominally a single batch.
Many papers say these problems were controlled. In reality, the process is difficult to control well. Randomizing the sequence can reduce the impact to some extent, but it rarely eliminates the quantitative inaccuracy caused by limited throughput and instrument drift.
Statistical models are not a magic exit
Suppose the analytical chemistry problems were all solved. The next layer is statistical analysis, and this is not much easier.
Which model should be used? Why that model rather than another? At present, these choices are often hard to test in a convincing way. It is difficult to say which model is best; in many cases, none is especially satisfying.
Some studies purchase thousands of standards to evaluate their methods. That helps, but it does not solve the central problem. Non-targeted analysis is designed to measure what is unknown, and no standard library can fully cover the unknown space.
Model complexity can be raised or lowered. Too much complexity risks overfitting; too little risks underfitting. There is nothing inherently wrong with trying hundreds of statistical or machine-learning models at once. The harder question is interpretation. Linear models and hierarchical models remain among the most interpretable options, but their predictive performance is often limited. Neural networks can be used, but interpretability becomes weaker. Faced with extremely tangled data, it is not surprising that clinical researchers often retreat to multivariable linear regression.
From mass-to-charge ratios to biological meaning
A related problem is QSPR, or quantitative structure–property relationship modeling. When metabolites or exposure-related compounds differ between groups, researchers usually want to infer chemical structures. Clinical studies often have defined endpoints, but environmental studies may not have clear groupings, or the groups may not support reliable effect prediction.
Effect-directed analysis can help from this angle, but the effect endpoint is still usually fixed. QSPR offers a way to predict multiple toxicity endpoints at once. Yet the bottleneck remains the same: converting a mass-to-charge signal into a reliable structure is still a tangled problem.
Multiple toxicity endpoints also imply multiple health models. That raises a larger question: is there a macro-model that can integrate several health models at the same time? Answering that cannot be done by one field in isolation. Collaboration is not just a nice ideal here; it is structurally necessary, because no single discipline has fully solved its own part of the problem.
Confounding is almost endless
Health-related research also faces the problem of effectively infinite confounders. Some are obvious: age, sex, race, and similar variables. Others may be ignored during modeling, or may not even be recognized as potential confounders.
Traditional studies often look for point-to-point correlations. Omics studies tend to look for one-to-many correlations. But the reality of health is many-to-many interaction. Controlled experiments are necessary, but when the data come from observational studies, the problem can become nearly unsolvable, constrained by the field of vision of the research community itself.
If only strong signals are followed, weak signals may be missed. But in this context, “strong” and “weak” are often determined by the instrument, not by biological importance. Many studies can tell a plausible story. Far fewer can answer a real question.
A field still learning where it can fail
The problems above are only a small fraction of the current obstacles. Progress on any one of them could overturn assumptions upstream and downstream. Standardized methods, reproducible workflows, and rapid integration with advances in basic research are all necessary.
Ten years from now, people may look back at today’s exposomics and wonder why so many resources were spent on studies that led nowhere. But that is part of how research works. We cannot know all of today’s mistakes in advance. What matters is recognizing that the problems are already visible.
To work in a new field at dawn is both fortunate and unfortunate. The fortunate part is that almost everyone starts from a similar line. The unfortunate part is that once the running begins, falling is almost guaranteed.